Genetic Treatments for Sickle Cell: When it comes to a devastating blood disorder, sometimes two wro
- May 1, 2016
- 7 min read
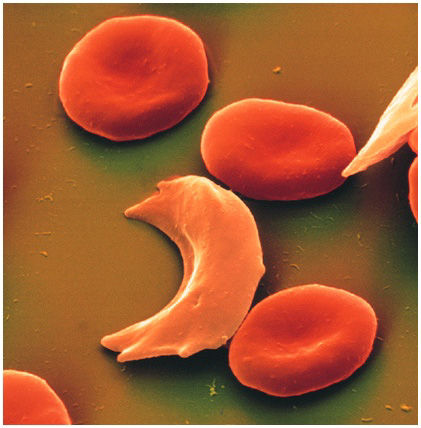
Ceniya Harris, age nine, of Boston should be a very sick little girl. Both her parents unknowingly passed her a copy of the genetic mutation for sickle-cell disease, a debilitating and sometimes fatal blood disorder. With a double dose of the mutant gene, Ceniya's body produces a defective kind of hemoglobin—the molecule in red blood cells that takes oxygen from the lungs and releases it into tissues throughout the body. The flawed hemoglobin molecules can deform the normally round blood cells into a crescent, or sickle, shape, leading the cells to clump together and hinder oxygen's passage into tissues. The subsequent physiological havoc, known as a sickle-cell crisis, is incredibly painful and frequently requires emergency treatment to prevent life-threatening strokes and organ failure.
Click here to read the story on ScientificAmerican.com
And yet the bouncy fourth grader, who is fond of sparkly shoes, is able to dance, participate in gymnastics and attend school without a hint of any health troubles. The secret to Ceniya's good fortune lies in a second genetic mutation she inherited—one that limits the aberrant curving of red blood cells. This unusual combination of genetic alterations means that she has yet to suffer a sickle-cell crisis, and her doctors believe that she will probably be protected from the effects of the defective hemoglobin for the rest of her life.
For decades physicians have known that a few children like Ceniya have unusual genetic mutations that counteract the effects of the sickle-cell flaw. Researchers would like to re-create their uncommon physiology in everyone with sickle-cell anemia. Though not technically a cure, the compensatory treatment would spare many of the 300,000 infants around the world who are born every year with sickle cell and who often do not live beyond childhood. It would also make life a lot easier for the more than 70,000 individuals living with the disease in the U.S., who, despite treatment that mitigates some of the most serious effects of the condition, often die in their 40s.
Investigators are now beginning to test such approaches, which depend on the precise alteration, or editing, of certain genes using new techniques in genetic engineering. (As will be addressed shortly, providing the compensatory mechanism should be easier to achieve than fixing the original sickle-cell genetic defect.)
“Gene-editing approaches have the potential to be game changing and really revolutionize the therapy,” says Lloyd Klickstein of health care company Novartis, which is among the firms and universities exploring new sickle-cell treatments.
WHAT DOESN'T KILL YOU
For a condition that causes life-threatening problems in childhood, sickle cell is surprisingly widespread. After all, if a mutation tends to kill people in childhood, you would expect few of the affected individuals to live long enough to mate and pass the trait to the next generation. The spread could be explained, however, if inheritance of just one copy of the mutation somehow protected people against a different threat to survival. In the late 1940s British scientist J.B.S. Haldane noted that inherited hemoglobin disorders are common in tropical areas where malaria is prevalent. He hypothesized that children born with a single mutated hemoglobin gene (which does not cause major problems) were somehow better able than their peers to fight off malaria and would survive to deliver the gene to their future children.
Subsequent studies supported Haldane's hypothesis, at least in part. Although individuals who carry a single sickle-cell gene actually can acquire malaria, they are less likely to die from the parasitic infection than those who do not have the mutation. How the altered hemoglobin confers this biological protection is still not entirely understood.
In contrast, researchers understand pretty well why two copies of the sickle-cell mutation can prove lethal. A molecule of hemoglobin is made up of four subunits: most commonly two identical proteins called alpha-globins and another pair of proteins known as beta-globins. Each of these subunits contains an iron-bearing structure, which, under normal circumstances, can grab on to or release a molecule of oxygen. Thus, each hemoglobin can carry up to four oxygen molecules. Individuals who inherit a single sickle-cell mutation produce one defective and one normal beta-globin; those who inherit sickle-cell genes from both parents produce only defective beta-globins.
When there is not enough oxygen around, these two defective beta-globins attach to each other. The connection is so tight that it causes the rest of the hemoglobin molecule to link up with other similarly affected hemoglobin molecules. The molecules end up forming long strands that distort the red blood cells into the sickle shape responsible for sickle-cell crises. Eventually the malformed molecules poke through the red blood cells in which they are found, like nails inside a plastic bag, says Matthew Heeney of the Dana-Farber/Boston Children's Cancer and Blood Disorders Center. The defective hemoglobin can puncture the cell, reducing its life span from the typical 120 days to fewer than 20 days. The body tries to replace these lost red blood cells, but if it cannot keep up, the resulting anemia causes its tissues to become oxygen-starved. The resulting tissue damage also triggers inflammation that can damage vessels and tissues.
LESSONS FROM NATURE
The only known cure for sickle-cell disease is bone marrow transplantation, which, in effect, provides a new circulatory system. But transplantation is expensive and requires a level of medical expertise that is unavailable in all but the wealthiest countries. Even there it is an option only for people who have an unaffected sibling with the right “tissue match.” As if that were not challenging enough, the procedure itself carries about a 5 to 10 percent risk of death, presenting parents with a terrible choice between risking their child's life and relieving his or her pain.
There is another situation, however, in which everyone with sickle-cell disease gets a temporary reprieve from its life-threatening effects: during development in the womb.
A fetus has a distinct kind of hemoglobin that binds very tightly to oxygen, allowing it to compete successfully with its mother's hemoglobin for oxygen in the placenta. Sometime early in an infant's first year after birth, the production of this fetal hemoglobin usually drops off, decreasing the amount of oxygen found in red blood cells. In a child who inherits the sickle-cell flaw from each parent, cells usually start to sickle several months after birth. And the march of symptoms begins.
Intriguingly, no one ever shuts off production of fetal hemoglobin entirely. Most adults—whether they have sickle-cell disease or not—produce about 1 percent fetal hemoglobin. In Ceniya's case, the gene that codes for fetal hemoglobin never received the message that it was no longer needed. Her hemoglobin remains 20 percent fetal, high enough to continue protecting her. The abundant oxygen it supplies to her red blood cells keeps her defective hemoglobin from behaving badly.
MAKING REPAIRS
The new idea for therapy would reawaken the fetal hemoglobin gene by disabling yet another gene—the one that essentially tells the fetal hemoglobin gene to stop working. How could giving sickle-cell patients the equivalent of a second genetic mutation be more practical than repairing the mutation that causes their condition in the first place? Because, at the moment, turning genes off is a lot easier than replacing the single mistake in the DNA molecule that causes the disease.
After decades spent studying the genetic underpinnings of sickle cell, Stuart Orkin, another researcher at Dana-Farber/Boston Children's, recently located the precise spot in the DNA of long-lived blood-making cells (stem cells) where a tiny snip would allow for indefinite manufacture of fetal hemoglobin. By inducing this mutation, Orkin and his colleagues have found a way to make two wrongs into a right. Sangamo BioSciences, in collaboration with Biogen, is gearing up to test a gene-editing technology using so-called zinc-finger scissors to make the snip that Orkin recommends. And Novartis's Klickstein says that, among other approaches, his company hopes to do the same with a different technique, known as CRISPR/Cas9.
Other firms are exploring the possibility that protective genes can be safely added after all. One such company, known as bluebird bio (based in Cambridge, Mass.), relies on a virus to deliver an antisickling gene that triggers production of healthy hemoglobin in blood-making stem cells. A year after a French 13-year-old with severe disease received the treatment, he was faring well, with no sickling events and no need for painkillers, says bluebird bio's chief medical officer David Davidson. The group has begun a 20-person study in the U.S. to explore the procedure further.
Both these paths are fraught with dangers, however. Patients need to undergo chemotherapy to wipe out most of the existing stem cells that make the wrong kind of hemoglobin so that there is room for new cells that make the right kind. Even though such treatment would be available to more people than are candidates for bone marrow transplants, it is so toxic in its own right that it can cause cancers to develop years after treatment. And virtually all patients will become infertile as a result. Again, a terrible decision for a parent to make on behalf of a child.
Global Blood Therapeutics in South San Francisco hopes to develop a drug that achieves the same benefits as gene therapy but without the side effects. The goal is to keep mutated hemoglobins from glomming on to one another—something they cannot do if they are holding on to an oxygen molecule. So the company is developing a pill, currently called gbt440, that binds oxygen to the alpha-globins longer than usual. Delaying their release of oxygen even just a bit prevents the beta-globins from getting close enough to connect—especially in the tiny capillaries where sickling occurs most often. Global Blood Therapeutics expects to begin a clinical trial later this year.
But as much as these therapies may make a difference for patients with sickle cell who live in wealthy countries, an entirely different solution is needed to help children elsewhere. “It's the undeveloped world that has the burden [of this disease],” says David G. Nathan, president emeritus of the Dana-Farber Cancer Institute and a leading sickle-cell researcher. For patients in the U.S., living with sickle cell is difficult, painful and terrifying; in the developing world, Nathan says, “it's a disaster.”






















Comments